Welcome to mBARq’s documentation!
Contents
Welcome to mBARq’s documentation!#
Transposon mutagenesis is a powerful technique that allows the identification of bacterial fitness factors under different environmental conditions. Recently, several studies have used barcoded transposon mutant libraries to increase the throughput of the experiments. mBARq allows easy processing and analysis of barcoded mutant libraries for any transposon construct (Tn5, mariner, etc).
Workflow#
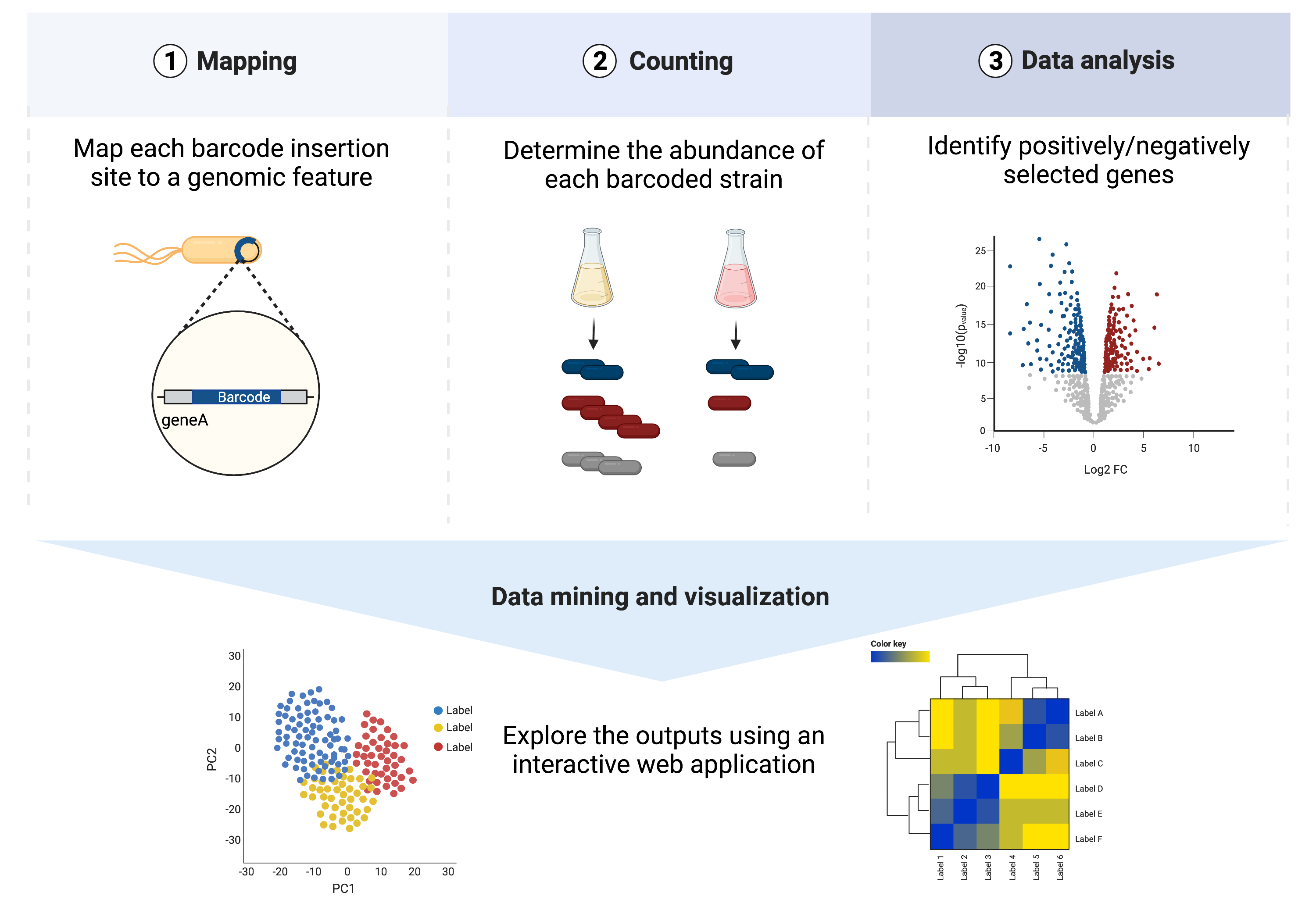
The main steps of the workflow involve:
Mapping of each barcode to the insertion location in the genome.
Profiling barcode abundances across samples.
Mutant fitness analyses.
Exploratory analysis using mBARq web app
Installation#
You will need conda to install and run mBARq.
Important
It is highly recommended that you install mamba as it greatly speeds up the environment creation. You can read more on its installation here.
Note
Some of the mBARq dependencies can only run on Linux of MacOS, as a consequence mBARq will also not run on a Windows system.
Option 1#
Download this environment file and run
mamba env create -f mbarq_environment_install.yaml
conda activate mbarq
mbarq --help
Option 2#
Clone the repository and create and activate the conda environment
git clone https://github.com/MicrobiologyETHZ/mbarq.git
cd mbarq
mamba env create -f mbarq_environment.yaml
conda activate mbarq
pip install -e .
mbarq --help
Quick Start#
Map each barcode to insertion location in the genome
mbarq map -f <library_R1.fastq.gz> -g <host.fasta> -a <host.gff> -l 100 \
-n LibraryName -tn B17N13GTGTATAAGAGACAG
Profile barcode abundances for each sample
mbarq count -f <sample.fastq.gz> -m <library_mapping_file.csv> \
-n ExperimentName -tn B17N13GTGTATAAGAGACAG
Merge barcode counts from multiple samples into the final table
mbarq merge -d <directory_with_count_files> -a locus_tag -n ExperimentName -o .
Identify enriched/depleted genes between treatments and control
mbarq analyze -i <count_file> -s <sample_data_file> -c <control_file> --treatment_column treatment \
--batch_column batch --baseline control
Walkthroughs:
Documentation: